Прямі серцево-судинні ефекти іНЗКТГ2 як одна з основ клінічної ефективності при серцевій недостатності: огляд експериментальних данихСеред кардіологів і суміжних фахівців інгібітори натрійзалежного котранспортера глюкози (НЗКТГ) 2 типу (іНЗКТГ2, гліфлозини) на сьогодні відомі тим, що знижують ризик комбінації смерті від серцево-судинних (СС) причин і госпіталізації з приводу серцевої недостатності (СН) у хворих на СН із цукровим діабетом (ЦД) або без нього [1, 2]. Зазначений СС ефект інгібіторів НЗКТГ2 (НЗКТГ2) є подвійно цікавим тому, що спочатку цю групу розробляли як протидіабетичні препарати, що пригнічують реабсорбцію глюкози з гломерулярного фільтрату в проксимальних звивистих канальцях нирки. Цей опосередкований нирками цільовий ефект іНЗКТГ2, безумовно, позитивно впливає на метаболічний, гормональний і гемодинамічний гомеостаз, але не може пояснити весь спектр СС ефектів іНЗКТГ2, оскільки дослідження інших препаратів із зіставним впливом на обмін глюкози або артеріальний тиск (АТ) не виявили таких виражених і швидких корисних СС ефектів, що спостерігалися при застосуванні іНЗКТГ2 [3]. Подальше вивчення ефектів іНЗКТГ2 in vitro показало, що ці препарати викликають значні зміни в ізольованих кардіоміоцитах [4, 5], які зазвичай не містять НЗКТГ2 [6]. Згодом дослідження продемонстрували наявність у іНЗКТГ2 прямих ефектів на різні клітини СС системи, зокрема на кардіоміоцити, ендотеліоцити, фібробласти і гладком'язові клітини, які також значною мірою позбавлені НЗКТГ2. Незабаром було встановлено, що значна кількість із цих прямих ефектів на клітини СС системи (далі – прямі СС ефекти) спрямовані на патогенетичні механізми розвитку й прогресування СН і діабетичної кардіоміопатії і тому можуть брати участь у реалізації зареєстрованих клінічних СС ефектів іНЗКТГ2 [7–9]. З метою аналізу наявної інформації про прямі СС ефекти іНЗКТГ2 Chen et al. виконали огляд літератури, що узагальнює поточні знання про прямий вплив іНЗКТГ2 на іонний гомеостаз та енергетичний статус, а також на такі процеси, як запалення, окислювальний стрес, проліферація, фіброз і обмін речовин у кардіоміоцитах, ендотеліоцитах, гладком’язових клітинах, фібробластах, тромбоцитах та ізольованих серцях тварин. На думку авторів, впливаючи на різні СС клітини, іНЗКТГ2 модулюють багато важливих базових клітинних механізмів, які доказово беруть участь у розвитку таких СС порушень, як гіпертрофія, СН, діастолічна дисфункція та аритмії, при цьому зазначені ефекти іНЗКТГ2 є взаємозалежними й можуть бути інтегровані в так званий серцевий натрієвий інтерактом (cardiac sodium interactome). Загалом до огляду Chen et al. увійшли 40 повнотекстових статей, опублікованих за період із 1975 по 2021 рік [10].
Іонний гомеостаз та іонні транспортери / обмінники 1. Кардіоміоцити та ізольовані серця. Інтерес до впливу іНЗКТГ2 на іонний гомеостаз пояснюється тим, що, за даними досліджень, підвищення концентрацій Na+ і Ca2+ у цитоплазмі (цитоплазматична концентрація, ЦПК) і зниження концентрації Ca2+ у мітохондріях (мітохондріальна концентрація, МК) кардіоміоцитів є факторами, що сприяють розвитку СН і серцевої смерті. Кілька досліджень виявили, що іНЗКТГ2 впливають на гомеостаз Na+ і Ca2+ в кардіоміоцитах. Зокрема, Hamouda et al. [5] показали, що дапагліфлозин у концентрації 1 мкМ знижував ЦПК Ca2+ під час систоли в ізольованих шлуночкових кардіоміоцитах у мишей зі стрептозоциніндукованим ЦД. Baartscheer et al. виявили, що короткочасна (<10 хв) або триваліша (3 год) інкубація здорових кардіоміоцитів кролика з емпагліфлозином у концентрації 0,2–1,0 мкМ знижувала ЦПК Na+, одночасно зменшуючи ЦПК Ca2+ [4]. Ці ефекти не залежали від глюкози, а тому не могли бути опосередковані ані через НЗКТГ 1 типу (НЗКТГ1), ані через НЗКТГ2. Подальші експерименти показали, що емпагліфлозин безпосередньо інгібує натрій-водневий обмінник (NHE)-1 у кардіоміоцитах кролика, сприяючи зниженню ЦПК Na+. Інгібувальну дію емпагліфлозину на NHE-1 нещодавно було підтверджено для клітин людського передсердя і міоцитів мишачих шлуночків [11]. Також нещодавно за допомогою методів поверхневого плазмонного резонансу було продемонстровано пряме зв’язування дапагліфлозину з NHE-1 [12]. Ефекти на внутрішньоклітинні концентрації Na+ можна вважати класовим ефектом іНЗКТГ2, оскільки і емпагліфлозин (1 мкМ), і дапагліфлозин (1 мкМ), і канагліфлозин (3 мкМ) знижували ЦПК Na+ та інгібували активність NHE-1 у здорових кардіоміоцитах мишей [13]. Проте не всі експерименти показали подібні результати, наприклад Chung et al. не змогли довести зниження ЦПК Na+ або пригнічення активності NHE-1 при впливі емпагліфлозину на ізольовані здорові серця миші, щура й морської свинки за допомогою методу ядерного магнітного резонансу [14]. Дані на підтримку ефекту іНЗКТГ2 на ЦПК Ca2+ ще більш неоднорідні, а відомості про вплив цих препаратів на МК Ca2+, хоча і багатообіцяючі, але нечисленні [10]. Проте зміни в іонному гомеостазі, викликані іНЗКТГ2, підтверджуються змінами в електрофізіології серця. Так, емпагліфлозин інгібував пізній компонент натрієвого струму (INaL) у кардіоміоцитах, отриманих у мишей із СН або з мутацією натрієвого каналу, до того ж емпагліфлозин, дапагліфлозин і канагліфлозин виявилися потужними й селективними інгібіторами INaL, спричиненого перекисом водню (H2O2). При цьому відомо, що інгібування INaL зменшує пролонгацію потенціалу дії та може знизити пов’язаний із нею ризик аритмій [15].

2. Ендотеліальні клітини. Chung et al. нещодавно показали, що емпагліфлозин (1 мкМ) також пригнічує активність NHE-1 і швидко знижує ЦПК Na+ в ендотеліоцитах коронарних артерій людини (HCAEC) і в ендотеліальних клітинах пупкової вени людини (HUVEC) [16], а також що емпагліфлозин (1 мкМ) пригнічує спричинену розтягуванням продукцію активних форм кисню (АФК) у HCAEC, що принаймні частково зумовлено інгібуванням NHE-1 [17]. Cappetta et al. також спостерігали інгібування NHE в HUVEC під впливом дапагліфлозину (1 мкМ) [18]. 3. Кардіофібробласти. У дослідженні Ye et al. [19] 16-годинна інкубація кардіофібробластів із дапагліфлозином (0,4 мкМ) пригнічувала індуковане ліпополісахаридами (ЛПС) збільшення синтезу матричної РНК (мРНК) NHE-1, при цьому дапагліфлозин специфічно інгібував зв'язування NHE-1 із білком теплового шоку 70 (Hsp70), не впливаючи на загальні рівні білка NHE-1 [19]. 4. Тромбоцити. Spigoni et al. [20] виявили, що 15-хвилинна попередня інкубація з емпагліфілозином (1 мкМ) або дапагліфілозином (1 мкМ) пригнічувала активацію тромбоцитів людини в спосіб, пов'язаний із NHE-1. Схожі результати спостерігалися в експерименті Lescano et al. [21], який показав, що іНЗКТГ2 зменшують ступінь активації тромбоцитів людини, особливо за присутності ендотеліальних факторів, таких як оксид азоту (NO) і простагландини. При цьому в жодному із цих досліджень у тромбоцитах не вдалося виявити ані мРНК НЗКТГ2, ані сам білок НЗКТГ2. Отже, результати більшості експериментів підтверджують, що іНЗКТГ2 пригнічують активність NHE-1 та інгібують INaL на плазматичній мембрані, що проявляється зниженням ЦПК Na+ і модуляцією ЦПК і МК Ca2+ в кардіоміоцитах. Той факт, що наявність зазначених ефектів спостерігалася не у всіх відповідних дослідженнях, дає змогу вважати, що деякі точки впливу іНЗКТГ2 досі невідомі. Крім того, можна припустити, що ці точки впливу пов'язані з патологічними станами, оскільки в більшості успішних експериментів прямі СС ефекти іНЗКТГ2 спостерігалися в умовах клітинного стресу [10].
Окислювальний стрес і запалення 1. Кардіоміоцити та ізольовані серця. Окислювальний стрес і запалення роблять значний і взаємозалежний внесок у розвиток і прогресування практично будь-якого СС захворювання. Запалення може прямо або опосередковано викликати гострий або хронічний окислювальний стрес через різні клітинні сигнальні шляхи за участю таких медіаторів, як цитоплазматична протеїнкіназа C (PKC) і кальцій, що активують джерела АФК, такі як нікотинамідаденіндинуклеотидфосфатоксидаза (NOX) та дихальний ланцюг перенесення електронів мітохондрій. Своєю чергою, окислювальний стрес є сильним модулятором запальної реакції, що діє через спричинену АФК експресію NLRP3 (кріопірин)-інфламмасоми та ядерного фактора каппа B (NF-kB) [22–24]. Згідно з отриманими даними, іНЗКТГ2 здатні моделювати ці процеси. Так, було показано, що в кардіоміоцитах тварин, які приймали їжу з високим вмістом жирів, 2-годинна інкубація з емпагліфлозином (1 мкМ) значно відновлювала знижені рівні Sestrin2 (регулятор антиоксидантних реакцій) і фосфорилювання активованої аденозинмонофосфатом протеїнкінази (AMPK; контролює енергетичний баланс клітини, за активації переводить клітину в енергозберігаючий стан) [25]. Однак у кардіоміоцитах тварин, які отримували стандартну їжу, подібного ефекту емпагліфлозину не виявлено, імовірно, через відсутність попереднього зниження рівня Sestrin2 і активності фосфорилювання AMPK. Крім того, було виявлено, що спричинене емпагліфлозином фосфорилювання AMPK стимулювало антиоксидантні реакції в мишей дикого типу, але не в мишей із нокаутом гена, що кодує Sestrin2. Ці дані свідчать про те, що емпагліфлозин може активувати AMPK і знижувати підвищені під впливом високого споживання жиру рівні АФК тільки в кардіоміоцитах, які перебувають у стані метаболічного стресу, і робить це за допомогою активації регулятора Sestrin2. В іншому дослідженні, проведеному на свіжоізольованих мишачих кардіоміоцитах, емпагліфлозин (0,5 мкМ) знижував рівні АФК, підвищені під впливом гіпоксії / реоксигенації [26]. Вираженість цього ефекту зменшувалася в присутності інгібітора АМРК (Сompound C), наводячи на думку про те, що зниження рівнів АФК під дією емпагліфлозину частково опосередковано активацією AMPK [26]. Крім того, у дослідженні на кардіоміоцитах із мишачих передсердь (HL-1) 8-годинна інкубація з емпагліфлозином (1 мкМ) призводила до інгібування індукованої ЛПС експресії фактора некрозу пухлини альфа (ФНП-α) та індуцибельної синтази оксиду азоту (iNOS) [27]. Зі свого боку Kolijn et al. [28] показали, що 1-годинна обробка емпагліфлозином (0,5 мкМ) м’язових волокон, ізольованих із біоптатів лівих шлуночків (ЛШ) пацієнтів із СН та збереженою фракцією викиду ЛШ, значно зменшила значення параметрів окиснення, що забезпечило краще фосфорилювання білків міофіламентів, зменшення жорсткості кардіоміоцитів і зниження рівнів цитокінів. Проте ці ефекти спостерігалися після обробки кардіоміоцитів, що робить їхню мембрану проникною для емпагліфлозину, тоді як відомостей, які б підтверджували поглинання іНЗКТГ2 неушкодженими серцевими клітинами, поки недостатньо. В ізольованих серцях мишей, попередньо оброблених емпагліфлозином (1 мкМ) та підданих 30-хвилинній ішемії та 40-хвилинній реперфузії, спостерігалося пригнічення окисного стресу й запальних процесів, що проявлялося, зокрема, зниженням активності каспази-1 (ініціатор запальної відповіді), зменшенням експресії білка, що взаємодіє з тіоредоксином (TXNIP, ендогенний інгібітор низки антиоксидантів), а також зниженням рівнів транскриптів численних маркерів запалення, таких як інтерлейкін (ІЛ)-18, ІЛ-1β і ФНП-α. Цікаво, що здатність емпагліфлозину запобігати запаленню, індукованому ЛПС, повністю пригнічувалася іонофором кальцію, тобто спричинене емпагліфлозином інгібування ЛПС-індукованого праймування NLRP3-інфламмасоми й продукції цитокінів, імовірно, опосередковане зниженням рівнів Са2+ [29]. Quagliariello et al. [30] продемонстрували, що в культурі клітин HL-1 24-годинна інкубація з емпагліфлозином (10–500 нМ) знижувала індуковану доксозазином (DOXO) експресію ІЛ-8, ІЛ-6, ІЛ1-β, лейкотрієну B4, мієлоїдного фактора диференціювання 88 (Myd88), NLRP3 і p65/ NF-kB. Більше того, у моделі гострого ушкодження міокарда перфузія ізольованих сердець мишей із додаванням 1 мкМ емпагліфлозину запобігала активації NLRP3-інфламмасоми й покращувала відновлення функцій після ішемії [29]. І, нарешті, у кардіоміоцитах H9C2 емпагліфлозин (1 мкМ) знижував індуковану ІЛ-1α активність матриксних металопротеїназ 2 і 9 та запобігав апоптозу [31]. Цікаво, що в дослідженні на тканинах правого і лівого передсердь людини та культивованих клітинах канагліфлозин (3 мкМ) знижував утворення АФК за допомогою інгібування шляху НЗКТГ1/AMPK/Rac1/ NOX, тоді як емпагліфлозин не виявив жодного ефекту [32]. 2. Ендотеліальні клітини. Уперше про прямі ендотеліотропні ефекти іНЗКТГ2 повідомили Gaspari et al. [33], які показали, що низькі концентрації дапагліфлозину (<5 нМ) пригнічують індуковану ФНП-α експресію мРНК NF-кB у клітинах HUVEC. В іншому дослідженні 15-хвилинна інкубація людських аортальних ендотеліальних клітин (HAEC) із клінічно значущими концентраціями канагліфлозину (але не емпагліфлозину та не дапагліфлозину) приводила до активації AMPK і водночас пригнічувала стимульовану ІЛ-1β адгезію промоноцитів U937, а також секрецію ІЛ-6 і моноцитарного хемоаттрактантного білка-1 (MCP-1) [34]. Ефекти низької дози емпагліфлозину (100 нМ) на спричинене гіперглікемією старіння нативних і свіжоізольованих ендотеліоцитів коронарних артерій свині показують, що цей іНЗКТГ2 знижує продукцію АФК, спричинену високим рівнем глюкози, і відновлює експресію eNOS і синтез NO за допомогою захисту від активації ендогенної системи ангіотензину [35]. Подальші експерименти показали, що емпагліфлозин запобігає експресії НЗКТГ1/2, спричиненій впливом високих рівнів глюкози на ендотеліоцити, а також зменшує поглинання глюкози за гіперглікемії, але не за нормоглікемії. Двогодинна попередня обробка 1 мкМ емпагліфлозину відновлювала корисний ефект ізольованих ендотеліоцитів мікросудин серця (CMEC) на здатність кардіоміоцитів до скорочення та розслаблення під час інкубації CMEC із ФНП-α [36]. Також емпагліфлозин пригнічував спричинене ФНП-α накопичення АФК у мітохондріях і цитоплазмі, що створювало умови для відновлення доставки NO, синтезованого в CMEC, до кардіоміоцитів (цей ефект спостерігався за відсутності змін у ступені експресії або фосфорилювання eNOS). Результати подальших досліджень показали, що таке зниження продукції АФК у CMEC можна приблизно на 30% пояснити інгібуванням NHE-1. У дослідженні Ortega et al. [37] 24-годинна попередня інкубація з емпагліфлозином (0,3 мкМ) пригнічувала спричинену ангіотензином II активацію сигнальних шляхів p38/мітогенактивована протеїнкіназа (MAPK) і p65/NF-kB у HAEC. Chung et al. також показали відновлення синтезу NO у поєднанні зі зменшенням продукції АФК під дією емпагліфлозину й дапагліфлозину в HCAEC, оброблених ФНП-α, без змін в експресії eNOS [38]. іНЗКТГ2 також знижували спричинену розтягненням продукцію АФК в HCAEC, і цей ефект був повністю відсутній за присутності інгібітора NHE1 карипориду або інгібітора NOX [17]. Крім того, нещодавно дослідники описали процес, що призводить до вироблення АФК в ендотеліальних клітинах у відповідь на запалення, а також механізм, за допомогою якого іНЗКТГ2 можуть пригнічувати цей процес [19]. Зокрема, у дослідженнях на HUVEC і HCEAC було виявлено, що вироблення АФК під впливом прозапального цитокіну ФНП-α опосередковується спричиненою ФНП-α активацією NHE-1 і, як наслідок, збільшенням внутрішньоклітинної концентрації Na+. Емпагліфлозин (1 мкМ) запобігав спричиненій запаленням продукції АФК за допомогою прямого інгібування NHE-1 і зниження внутрішньоклітинного рівня Na+. Додатковим шляхом зниження продукції АФК в ендотеліоцитах під дією іНЗКТГ2 може бути вплив на шлях ангіотензину II, який також може бути пов’язаний з інгібуванням NHE-1 [39]. 3. Гладком’язові клітини. У дослідженні Sukhanov et al. на культивованих клітинах гладких м'язів аорти людини емпагліфлозин (1 мкМ) знижував вираженість індукованого ІЛ17α окислювального стресу, пригнічував експресію NLRP3, активацію каспази-1, секрецію ІЛ-1β та ІЛ-18, частково за рахунок інгібування НЗКТГ2 [40]. Ці автори повідомили про наявність НЗКТГ2 у цих гладком’язових клітинах і про те, що експресія НЗКТГ2 збільшувалася під дією ІЛ-17α. 4. Кардіофібробласти. 16-годинна попередня інкубація з дапагліфлозином (≥0,3 мкМ) забезпечувала пригнічення експресії NRLP3, ФНП-α і каспази-1 в оброблених ЛПС свіжоізольованих кардіофібробластах зі здорових сердець мишей C57BL/6J, при цьому спостережуваний ефект був AMPK-залежним [41]. У сукупності ці дані показують, що іНЗКТГ2 безпосередньо зменшують активність запалення та/або тяжкість окислювального стресу в різних типах клітин за різноманітних патологічних станів, пов'язаних зі споживанням великої кількості жирів, гіпоксією / реоксигенацією, ішемією / реперфузією, впливом ЛПС, DOXO, ІЛ-1α, ІЛ-1β, ФНП-α, ІЛ-17α, ангіотензину II, гіперглікемії чи генератора окислювального стресу. У здорових клітинах ефекти іНЗКТГ2, спрямовані проти запального процесу та окислювального стресу, виражені значно слабкіше. Питання про те, якою мірою залежність ефектів іНЗКТГ2 від стресу пояснюється спричинюваною стресом експресією НЗКТГ1/2 та/або активацією NHE-1/INaL, потребує подальшого вивчення [10].
Метаболізм 24-годинна інкубація з 1 мкМ емпагліфлозину збільшувала експресію транспортера глюкози 1 (GLUT 1, забезпечує базальний рівень поглинання глюкози), але не GLUT4 (інсулінзалежний переносник глюкози), в ізольованих кардіоміоцитах із сердець пацієнтів із термінальною стадією СН, а також із сердець здорових мишей та мишей із СН, індукованою поперечним перетягуванням аорти (ТАС-миші) [42]. Також емпагліфлозин збільшував поглинання 2-дезоксиглюкози в ізольованих кардіоміоцитах TAC-мишей після періоду голодування (без подачі глюкози). У дослідженні на ізольованих серцях мишей із ЦД 2 типу, присвяченому оцінці метаболічних ефектів 35-хвилинної перфузії емпагліфлозину (1 мкМ) на метаболізм 13C-глюкози або 13C-пальмітату [43], емпагліфлозин зменшував утворення лактату із 13C-глюкози й збільшував утворення α-кетоглутарату із 13C-пальмітату. Зникнення цих змін у присутності інгібітора NHE-1 карипориду дає змогу припустити, що ці прямі метаболічні ефекти емпагліфлозину були опосередковані інгібуванням NHE-1 [39]. Загалом, є підстави вважати, що прямі метаболічні ефекти іНЗКТГ2 на серце можуть лише злегка змістити метаболізм із використання глюкози на окислення жирних кислот. Проте наразі проводять дослідження прямих СС ефектів іНЗКТГ2 в умовах тривалішого (понад 1 годину) впливу на патологічно змінені серця, у яких, як очікується, ефекти іНЗКТГ2 можуть бути продемонстровані чіткіше [10].
Серцева функція Ізольоване ex vivo перфузоване серце є ідеальною моделлю для вивчення прямих СС ефектів іНЗКТГ2, оскільки дає змогу відстежувати функцію неушкодженого серця, яке б'ється, під час оброблення іНЗКТГ2 за відсутності змін АТ, коронарного кровотоку, а також рівнів субстратів або гормонів, які доставляються в серце. Зібрати таку інформацію in vivo неможливо через виражений ефект іНЗКТГ2 на нирки зі стимуляцією діурезу, натрійурезу й глюкозурії. Під час нормоксичної перфузії серця збалансованою фізіологічною сумішшю важливих субстратів введення іНЗКТГ2 у концентрації 1–3 мкМ не впливало на жоден параметр функції серця, зареєстрований у серцях здорових або хворих на ЦД 2 типу мишей [13, 43, 44]. Відповідно до передбачуваного ефекту іНЗКТГ2 на NHE-1, інгібітор NHE-1 карипорид у цих умовах також не впливав на роботу серця [44], і це відповідає гіпотезі про низьку активність NHE-1 під час нормоксичних станів із нейтральним значенням pH (7,4), тобто в здоровому (патологічно незміненому) серці. Однак при створенні ішемічних умов іНЗКТГ2 справді впливали на роботу серця, затримуючи розвиток контрактури під час глобальної ішемії та зберігаючи скоротливу функцію під час регіональної ішемії [44, 45]. Більшість досліджень також показали покращення функції серця в присутності 1 мкМ емпагліфлозину після закінчення періоду ішемії [14, 46]. Отже, згідно з усталеною думкою, іНЗКТГ2, хоча й не мають впливу на функцію здорового серця, можуть покращувати функцію серця під час ішемії-реперфузії, тобто прямі СС ефекти іНЗКТГ2 здебільшого стають очевидними у хворому серці.
Обговорення Поява іНЗКТГ2 суттєво змінила ситуацію у сфері фармакотерапії ЦД і СН завдяки послідовним захисним ефектам цих препаратів на серце й нирки, що підтверджується клінічними наслідками в цілій низці великих клінічних досліджень [1, 2]. Так, згідно з нещодавнім метааналізом, який включив одинадцять досліджень з оцінкою СС наслідків і більш ніж 75 тис. пацієнтів, лікування іНЗКТГ2 у хворих на кардіометаболічні й ниркові захворювання призвело до стійкого зниження ризику комбінації СС смерті та госпіталізації з приводу СН, надійного зниження ризику подій, пов'язаних із СН, а також до помірного зниження СС смертності, загальної смертності та ризику серйозних несприятливих СС подій [47]. Загальновизнано, що настільки виражені корисні клінічні ефекти не можуть бути пояснені одним єдиним механізмом дії, націленим на НЗКТГ2, й, імовірно, зумовлені поєднанням цільових (ниркових) і нецільових ефектів іНЗКТГ2. Цільові ниркові ефекти включають стимуляцію глюкозурії, діурезу, натрійурезу і покращення функції нирок, що призводить до таких системних ефектів, як зниження АТ, поліпшення співвідношення «глюкоза/інсулін», підвищення синтезу еритропоетину, покращення взаємодії між нирками та серцем, а також оптимальніший перерозподіл рідини й натрію в організмі [8, 48, 49]. Вивчення нецільових і прямих ефектів іНЗКТГ2 перебуває на ранньому (експериментальному) етапі, але вже стає зрозумілим, що, впливаючи на різні типи клітин СС системи, іНЗКТГ2 модулюють багато важливих механізмів, які беруть участь у розвитку й прогресуванні серцевих порушень і захворювань, зокрема гіпертрофії, СН, діастолічної дисфункції та аритмії. Ця модуляція особливо очевидна в експериментах із впливом на клітини різноманітних патологічних медіаторів, наприклад запальних (цитокіни, ЛПС) та метаболічних (гіперглікемія, високий вміст жирів, низький вміст кисню, ішемія, ацидоз) стресових факторів, у яких додавання іНЗКТГ2 зменшувало вираженість спричинених патологічних змін іонного гомеостазу, окисно-відновлювального статусу, метаболізму та функції серця. Малоймовірно, що іНЗКТГ2 змінює ці процеси окремо і незалежно один від одного. Найімовірніше, ці чутливі до іНЗКТГ2 процеси пов’язані через спільний «вузол», на який впливають іНЗКТГ2, а саме через внутрішньоклітинний інтерактом, у центрі якого перебувають концентрації іонів Na+ (рис.) [10].
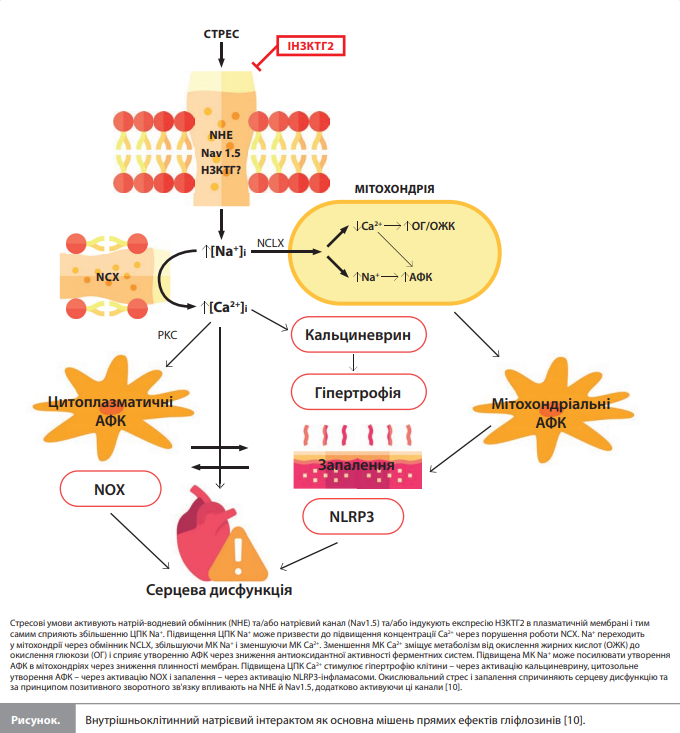
Внутрішньоклітинна концентрація Na+ як вузлова точка серцевого інтерактома, на яку впливають прямі СС ефекти іНЗКТГ2. Майже всі клітини в організмі характеризуються високим градієнтом натрію на клітинній мембрані (різниця концентрацій: близько 10 мМ усередині клітини і 135–145 мкМ – поза клітиною), який підтримується енергоємним натрій-калієвим насосом. Натрієвий градієнт необхідний для реалізації різних важливих фізіологічних процесів – від створення потенціалу дії в збудливих клітинах до полегшення клітинного обміну різними молекулами через Na-котранспортери / обмінники [10]. Зміни концентрацій Na+ можуть діяти як найважливіший сигнал, що регулює багато клітинних функцій, і мають прямі наслідки, наприклад, для транспорту Ca2+, розвитку окислювального стресу, регуляції імунних реакцій і запалення, функції мітохондрій. Підвищена внутрішньоклітинна концентрація Na+ є типовою характеристикою клітин серця за наявності СН і ЦД, при цьому збільшення концентрацій Na+ за механічного або метаболічного перевантаження пояснюється підвищеною активністю NHE, NCX, збільшенням INaL та/або активацією НЗКТГ 1 і 2 типів [50–55]. Збільшення концентрацій Na+ може запустити низку низхідних сигнальних шляхів, які доказово активні при СН. Так, підвищена внутрішньоклітинна концентрація Na+ викликає збільшення МК Na+ за рахунок виведення мітохондріального Ca2+ через мітохондріальний натрій-кальцієвий обмінник (NCLX). Збільшення МК Na+ зменшує плинність внутрішньої мембрани мітохондрій і тим самим сприяє виробленню мітохондріальних АФК [56]. Зниження МК Ca2+ перешкоджає утворенню відновленого нікотинамідаденіндинуклеотиду (НАДН) у циклі трикарбонових кислот, що призводить до дефіциту енергії та зниження утворення нікотинамідаденіндинуклеотидфосфату (НАДФ), необхідного для антиоксидантної активності [57]. Крім того, було показано, що підвищена ЦПК Na+ в кардіоміоцитах спричиняє зсув метаболізму з розщеплення жирних кислот на розщеплення глюкози через зменшення МК Ca2+ та/або збільшення ЦПК Ca2+ [58, 59]. Дефіцит енергії, зниження окислювальної здатності, посилення окислювального стресу, стимуляція гліколізу й послаблення метаболізму жирних кислот є типовими характеристиками СН, спричиненої артеріальною гіпертензією або перенесеним інфарктом міокарда [60–62]. Зниження окислювальної здатності, імовірно, спричинене не зменшенням постачання кисню або ішемією, а метаболічним ремоделюванням, оскільки при СН надходження кисню до міокарда не обмежується [63–65]. Згодом зростання ЦПК Ca2+ продовжується, що зумовлено інгібуванням натрій-кальцієвого обмінника в сарколемі (NCX). Це може спричинити цілу низку несприятливих змін, зокрема гіпертрофію серця (за допомогою активації кальційзалежної фосфатази кальциневрину [66]), а також активацію НАДФ-оксидази, що стимулює синтез АФК [67]. Зростання концентрації іонів Ca2+ поряд зі збільшенням цитозольної та мітохондріальної продукції АФК може активувати NLRP3-інфламмасому, що призводить до утворення прозапального ІЛ-1β [68], який може індукувати вироблення таких медіаторів запалення, як ІЛ-6 та ФНП-α. АФК і запальні медіатори потім можуть активувати NHE-1 сарколеми або посилювати INaL, що додатково сприяє збільшенню концентрацій Na+ [16, 69]. Результатом окислювального стресу також може стати окиснення Ca2+/ кальмодулінзалежної кінази IIδ (CaMKII), яка збільшує INaL та активує NHE-1, що призводить до подальшого перевантаження клітини Na+ і Ca2+ [70, 71]. Підвищена ЦПК Ca2+ також сприяє розвитку небезпечних для життя серцевих аритмій. Отже, передбачається, що за патологічних станів серця, для яких характерні запалення, окислювальний стрес і перевантаження іонами Na+ і Ca2+, наприклад, за СН, зазначені механізми підсилюють один одного за допомогою механізму позитивного зворотного зв'язку, що сприяє прогресуванню захворювання. Результати представлених експериментальних досліджень у сукупності показують, що іНЗКТГ2 безпосередньо пригнічують активність перерахованих механізмів у різних клітинах СС системи. Імовірно, іНЗКТГ2 гальмують позитивний зворотний зв'язок, що активується під час хворобливих станів, за допомогою пригнічення транспортерів Na+ у клітинній мембрані [10]. Суперечливість результатів досліджень прямих СС ефектів іНЗКТГ2. Як видно з наведених вище результатів експериментальних досліджень, для низки досліджуваних змінних були отримані суперечливі оцінки змін під дією іНЗКТГ2. Ці розбіжності результатів, отриманих у різних лабораторіях, можуть пояснюватися відмінностями в умовах експериментів і/або експериментальних моделях, які визначають: 1) присутність / активність пропонованих мішеней іНЗКТГ2 (NHE-1, Nav1.5, НЗКТГ); 2) можливість модулювання пропонованих результуючих змінних (NO, АМФ, Na+, Ca2+, АФК); 3) достатність чутливості застосованих методів для виявлення відносно невеликих коливань змінної [10]. Як приклад можна навести особливість взаємодії між іНЗКТГ2 і натрієвим каналом Nav1.5, що підсилює INaL, яка полягає в тому, що іНЗКТГ2 може впливати на цей канал тільки після його окислення під дією Н2О2 [15]. Також було виявлено, що ефекти іНЗКТГ2 на активність NHE-1 стають більш вираженими за нижчого значення pH [72], тобто за умов закислення, що пояснює, наприклад, результати експерименту, в якому інгібування NHE-1 емпагліфлозином спостерігали лише за наявності інгібітора натрій-калієвого насоса, який спричиняє ацидоз [73]. Крім того, NHE має низьку активність у здоровому міокарді, яка істотно підвищується, наприклад, за СН, так що зниження концентрацій натрію за допомогою інгібування NHE (або впливу іНЗКТГ2) набагато помітніше і легше виявляється в кардіоміоцитах, виділених із серця із СН, порівняно зі здоровими серцями [51]. Нарешті, нещодавно було виявлено, що патологічні стани, наприклад постінфарктний період, можуть стимулювати експресію НЗКТГ2 у різних органах, зокрема в серцевій тканині [39, 74, 75]. Транспорт глюкози через НЗКТГ2 відбувається одночасно з транспортом Na+ і тому також призводить до збільшення внутрішньоклітинних концентрацій Na+. У сукупності ці процеси призводять до того, що в умовах стресу клітини стають більш чутливими до впливу іНЗКТГ2, що ще раз підкреслює важливість умов експерименту. Що стосується чутливості результуючих змінних до модуляції досліджуваним препаратом, тут важливу роль відіграють метаболічні відмінності в різних клітинах або моделях, які визначають базовий фізіологічний стан, а отже, чутливість досліджуваної біологічної системи до змін, спричинених лікуванням іНЗКТГ2 [68, 76]. Наприклад, можливість посилення сигналізації AMPK у кардіоміоцитах тварин під дією іНЗКТГ2 залежить від характеру харчування [26] та інших чинників, що визначають метаболічний баланс між гліколізом та активністю мітохондрій і, отже, чутливість AMPK до модуляції [76]. Ефекти іНЗКТГ2 на активність eNOS теж спостерігалися не в усіх експериментах, що також може бути пов'язано з особливостями механічних і метаболічних умов, що застосовуються до ендотеліоцитів в окремих дослідженнях. Наприклад, високий рівень глюкози в живильному середовищі спричиняє особливу форму глікозилювання eNOS, відому як O-GlcNAcylation, що унеможливлює збільшення активності ферменту [77]. Нарешті, відмінності в чутливості методик вимірювання, що використовуються в різних лабораторіях, також можуть впливати на результати досліджень. Наприклад, у деяких експериментах іНЗКТГ2 спричиняв невеликі зниження рівнів Na+ (<2 мМ) при низьких абсолютних концентраціях іона [4, 13], які неможливо виміряти за допомогою стандартного методу флуоресцентного аналізу, але можна оцінити за допомогою специфічного флуоресцентного зонда [78].
Висновок Отже, хоча необхідно проводити додаткові дослідження, уже зараз можна сказати, що в іНЗКТГ2 є прямі ефекти на клітини СС системи, особливо виражені під час захворювань, і що ці ефекти опосередковані зміною внутрішньоклітинних концентрацій натрію. При цьому важливо відзначити, що, хоча СС ефекти іНЗКТГ2 спостерігалися не у всіх експериментах, вони майже завжди були очевидні в умовах, що відповідають стресу або захворюванню. Це дає змогу використовувати інформацію про клітинні ефекти іНЗКТГ2 для розуміння основних механізмів дії цих препаратів у клінічних умовах, коли іНЗКТГ2 використовують тільки у хворих людей [10].
Список літератури знаходиться в редакції
Автор огляду Микола Горін Medicine Review 2024; 7 (80): 24 |
Корисні посилання






|
|
Інформація, розміщена на сайті, призначена тільки для професіоналів охорони здоров'я та не може бути використана як інструкція для самолікування. |
Головна | Про видання | Поточний номер | Архів номерів | Новини | Правова інформація
Medicine Review © 2008—2026. Усі права захищені.
|
мапа сайту корисні посилання |
