Перша настанова ESC із ведення кардіоміопатій (2023): основні аспекти документаСеред практичних рекомендацій, представлених Європейським товариством кардіологів (ESC) на щорічному конгресі в серпні 2023 р., особливе місце посідає настанова з ведення кардіоміопатій (КМП), публікація якої, на думку фахівців, є найважливішою віхою в лікуванні цієї групи захворювань [1]. Дотепер доступні рекомендаційні ресурси були обмежені узгодженими документами, а також настановою з ведення пацієнтів із гіпертрофічною КМП (ГКМП) [2], тоді як новий документ ESC є першою всеохоплюючою збіркою рекомендацій, присвяченою КМП як загалом, так і за окремими формами кардіоміопатій [3]. Ключові моменти першої настанови ESC щодо ведення хворих на КМП стисло представлені в цьому огляді.
Класифікація КМП Насамперед, у настанові презентовано нову класифікацію КМП, засновану на морфологічних і функціональних характеристиках, яка покликана спростити термінологію, водночас забезпечивши концептуальне підґрунтя для діагностики й лікування. Ця класифікація передбачає такі фенотипи КМП (рис.) [3]: 1. ГКМП, що характеризується гіпертрофією лівого шлуночка (ЛШ) (тобто збільшенням товщини стінки або маси ЛШ), яка не пояснюється винятково аномальним навантаженням на ЛШ (наприклад, унаслідок артеріальної гіпертензії, клапанної хвороби або вроджених вад серця); 2. Дилатаційна кардіоміопатія (ДКМП), що характеризується дилатацією і глобальною або регіональною систолічною дисфункцією ЛШ, які не пояснюються винятково аномальним навантаженням на ЛШ або наявністю ішемічної хвороби серця. Дилатація і дисфункція правого шлуночка (ПШ) також можливі, але не обов’язкові для постановки діагнозу ДКМП. Якщо зміни в ПШ переважають, слід розглянути діагноз «аритмогенна КМП ПШ» (див. нижче);. 3. Недилатаційна кардіоміопатія ЛШ (НДКМП ЛШ) – нова форма захворювання (замінює запропоновану раніше в узгодженому документі ESC гіпокінетичну недилатаційну КМП), яка характеризується наявністю неішемічного фіброзу / рубцювання або жирового заміщення міокарда ЛШ (незалежно від наявності глобального або регіонального порушення скоротливості стінок ЛШ) або наявністю ізольованої глобальної гіпокінезії ЛШ без фіброзу / рубцювання міокарда; 4. Аритмогенна КМП ПШ (АКМП ПШ), що характеризується наявністю переважної гіпертрофії та/або дисфункції ПШ у поєднанні з гістологічними та/або електрокардіографічними змінами, що відповідають опублікованим критеріям АКМП ПШ; 5. Рестриктивна кардіоміопатія (РКМП), що характеризується наявністю рестриктивної патофізіології ЛШ та/або ПШ за наявності нормальних або зменшених діастолічних об’ємів (одного або обох шлуночків), нормальних або зменшених систолічних об’ємів і нормальної товщини стінок шлуночків. Також характерне збільшення розмірів обох передсердь. Робоча група підкреслює, що в одній сім'ї можуть співіснувати різні фенотипи КМП і що в ході розвитку захворювання в окремого пацієнта можливий перехід від одного фенотипу КМП до іншого. Відповідно, експерти вважають, що обґрунтованою тактикою під час вибору номенклатури й методів діагностики КМП є орієнтація на фенотип, що переважає на момент обстеження пацієнта. Додатково, у розділі, присвяченому класифікації КМП, замість терміну «некомпактність ЛШ» рекомендується використовувати термін «підвищена трабекуляція міокарда» (зокрема за тимчасового характеру цього феномена або явного його дебюту в дорослому віці), яку експерти вважають скоріше фенотиповою ознакою, аніж проявом КМП як такої, оскільки вона може бути наявною у представників здорової популяції (спортсмени, вагітні жінки тощо). Крім того, в настанові не рекомендується відносити до КМП синдром Такоцубо.
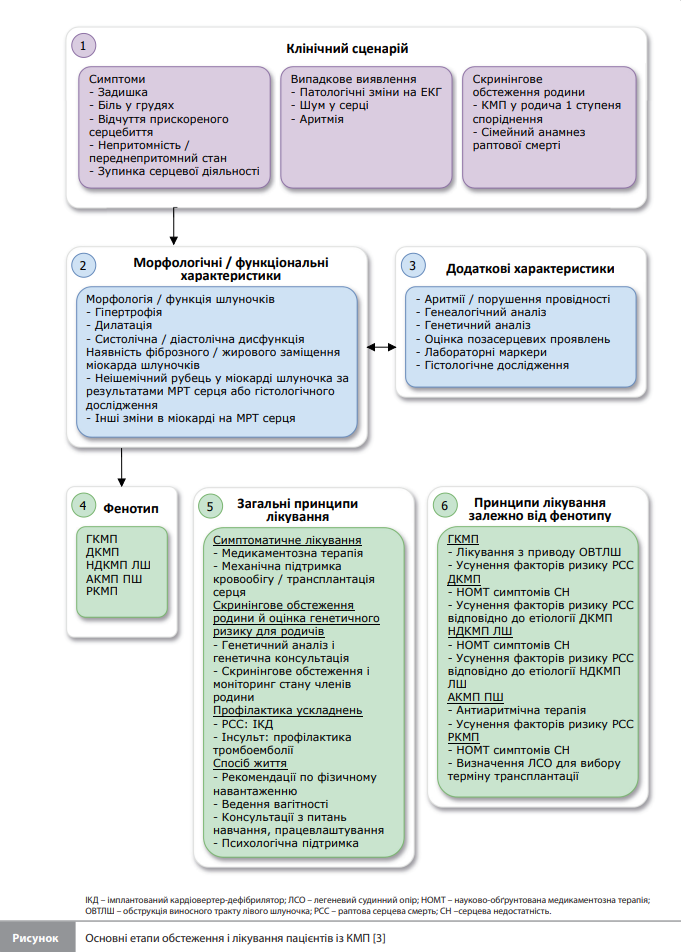
Діагностика КМП У настанові запропоновано системний підхід щодо визначення фенотипу КМП і постановки етіологічного діагнозу, водночас підкреслюється важливість побудови генеалогічного дерева на глибину до трьох поколінь, оцінювання серцевих і екстракардіальних ознак / симптомів, результатів електрокардіограми (ЕКГ), холтерівського моніторування ЕКГ, аналізів крові та даних, отриманих за допомогою методів мультимодальної візуалізації. Також рекомендується вимірювання рівнів серцевого тропоніну та N-термінального фрагмента попередника натрійуретичного пептиду, оскільки вони можуть мати діагностичне й прогностичне значення. При обстеженні пацієнтів дитячого та підліткового віку слід використовувати аналогічний підхід з урахуванням іншого профілю етіології КМП у дітей віком до року. Серед методів візуалізації провідна роль відводиться магнітній резонансній томографії (МРТ) серця з контрастом, яку рекомендується використовувати в усіх пацієнтів під час постановки діагнозу (рекомендація класу I, B) і розглядати як метод оцінювання під час повторних обстежень із частотою один раз на 2–5 років (IIa, C). Крім того, представлено рекомендацію класу IIa, B, що закликає розглядати можливість проведення МРТ серця членам сім'ї хворого на КМП, які мають пов'язані з КМП генетичні зміни без клінічних проявів КМП, для раннього виявлення характерних для КМП змін у серці. Іншими методами візуалізації, які можуть бути корисними на етапі первинної діагностики (для визначення фенотипу КМП і виключення фенокопій), стратифікації ризику, оцінки прогнозу й подальшого спостереження, є ехокардіографія (ЕхоКГ), зокрема з навантаженням, комп'ютерна томографія (КТ) коронарних артерій, сцинтиграфія кісток і позитронно-емісійна томографія, поєднана з КТ [3]. Серед методів обстеження пацієнтів із підозрою на КМП велике значення надається генетичному тестуванню, оскільки його результати мають діагностичну, прогностичну й терапевтичну цінність при різних КМП. Із повним переліком генів, доведено й можливо пов'язаних із розвитком різних КМП, можна ознайомитися в узгодженому документі EHRA від 2022 року [10]. Показанням до генетичного тестування є потенційний вплив його результатів на ведення пацієнта або оцінку членів сім'ї (I, B). Крім того, підкреслюється роль молекулярної аутопсії в посмертній діагностиці КМП [3]. Значна увага приділена стандартизації процедури спостереження за членами сім'ї хворого на КМП, яке має тривати до досягнення 50-річного віку для очевидно спорадичних випадків (негативні результати генетичного тестування і відсутність інших членів сім'ї з КМП) i бути довготривалішим, якщо в сім'ї є більше одного члена з КМП (для випадків із виявленою мутацією – клас рекомендації І, В, для випадків без виявленої мутації – IIa, C) [3].
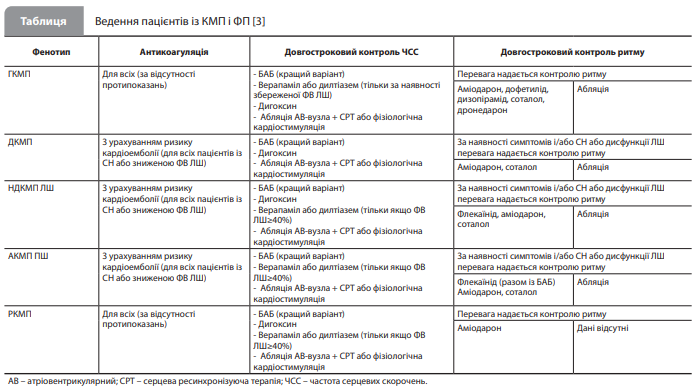
Загальні принципи терапії КМП У настанові детально описано загальні принципи терапії, застосовні до всіх КМП, у тому числі схвалені методи лікування серцевої недостатності (СН), передсердних (фібриляція і тріпотіння) і шлуночкових аритмій, а також методи апаратної терапії. При цьому зазначено, що підхід до лікування СН має ґрунтуватися не на фенотипі КМП, а на проявах СН, зокрема на функції ЛШ. Як наслідок, рекомендації з лікування СН зі зниженою фракцією викиду (ФВ) ЛШ застосовні переважно до фенотипів КМП зі зниженою функцією ЛШ, зокрема до генетично зумовленої ДКМП і НДКМП ЛШ, а рекомендації з лікування СН зі збереженою ФВ ЛШ – переважно до необструктивної ГКМП і РКМП. Слід зазначити, що в рекомендаціях передбачено можливість використання методів лікування СН у безсимптомних пацієнтів із ранніми стадіями ДКМП або НДКМП ЛШ для запобігання дилатації та дисфункції шлуночків унаслідок несприятливого ремоделювання міокарда (IIb, C). Щодо серцевих аритмій, то за наявності фібриляції або тріпотіння передсердь антикоагулянти рекомендують усім пацієнтам із ГКМП і амілоїдозом серця (I, B) або РКМП (IIa, C), незалежно від ризику інсульту / тромбоемболії (будь-яка оцінка за шкалою CHA2DS2-VASc), а також усім пацієнтам із ДКМП, НДКМП ЛШ і АКМП ПШ з оцінкою за CHA2DS2-VASc ≥2 для чоловіків і ≥3 для жінок (I, B). Особливий акцент зроблено на досягненні контролю серцевого ритму за допомогою антиаритмічних препаратів і процедури катетерної абляції (табл.) [3]. Інформація в розділах, присвячених шлуночковим аритміям і апаратній терапії з використанням імплантованих кардіовертерів-дефібриляторів (ІКД), ґрунтується на тексті настанови ESC із ведення пацієнтів зі шлуночковими аритміями й профілактики раптової серцевої смерті від 2022 року [11].
Рекомендації щодо окремих фенотипів ГКМП 1. ГКМП. У розділі, присвяченому ГКМП, значна увага приділяється новим відомостям і розробкам; із більшості інших питань автори посилаються на відповідні рекомендації ESC від 2014 року [2]. Одним із найбільш значущих нововведень є представлення мавакамтена (інгібітор АТФази серцевого міозину) як препарату для другої лінії терапії симптоматичної обструктивної ГКМП. Дія мавакамтена заснована на інгібуванні процесу утворення поперечних містків між актином і міозином, що зменшує скорочувальну здатність і покращує енергетичний стан міокарда. У нещодавно опублікованому дослідженні EXPLORER-HCM (Clinical Study to Evaluate Mavacamten in Adults with Symptomatic Obstructive Hypertrophic Cardiomyopathy) мавакамтен зменшував градієнт між порожниною і виносним трактом ЛШ (ВТЛШ) і покращував переносимість фізичних навантажень порівняно з плацебо в пацієнтів із ГКМП і симптоматичною обструкцією ВТЛШ (ОВТЛШ) (II–III функціональний клас [ФК] за класифікацією Нью-Йоркської кардіологічної асоціації [NYHA] і ФВ ЛШ >55%). Крім того, у 27% пацієнтів, які приймали мавакамтен, градієнт у ВТЛШ знизився до <30 мм рт. ст., а ФК за NYHA покращився до I [4]. Препарат добре переносився і демонстрував гарний профіль безпеки; лише в невеликої групи пацієнтів розвинулася минуща систолічна дисфункція ЛШ, яка зникла після тимчасового припинення приймання препарату. В іншому дослідженні, VALOR-HCM (A Study to Evaluate Mavacamten in Adults With Symptomatic Obstructive HCM Who Are Eligible for Septal Reduction Therapy), яке проводили за участю дорослих пацієнтів з обструктивною ГКМП, скерованих на септальну редукційну терапію (SRT) у зв'язку з некурабельними симптомами, мавакамтен значно зменшив частку пацієнтів, які відповідали критеріям проведення SRT, через 16 і 32 тижні лікування [5, 6]. Невеликі дослідження з проведенням МРТ серця і ЕхоКГ дають змогу припустити, що мавакамтен може також сприяти позитивному ремоделюванню міокарда зі зменшенням маси міокарда, товщини стінок ЛШ і об'єму лівого передсердя [7–9]. За відсутності прямих порівнянь експерти ESC не змогли рекомендувати використання інгібіторів АТФази серцевого міозину як терапію першої лінії, але визнали докази достатньо переконливими, щоб розглядати можливість застосування мавакамтену в дорослих симптоматичних пацієнтів з ОВТЛШ у спокої або внаслідок провокації в ситуаціях, коли оптимальна медикаментозна терапія бета-блокаторами (БАБ) або антагоністами кальцію (АК) неефективна або погано переноситься, з призначенням мавакамтену на додачу до БАБ або АК (IIa, A). Підвищення дози препарату до максимальних 15 мг слід виконувати під контролем ЕхоКГ. У пацієнтів із протипоказаннями або непереносимістю БАБ, АК або дизопіраміду інгібітори АТФази серцевого міозину можуть використовуватися в режимі монотерапіі (IIa, B) [3]. У підрозділі, присвяченому запобіганню РСС за ГКМП, детально розглядається питання застосування ІКД для первинної та вторинної профілактики РСС. Клас рекомендації щодо застосування ІКД для первинної профілактики залежить від рівня ризику згідно з балом за шкалою оцінювання 5-річного ризику РСС за ГКМП (HCM-risk scores): у групі низького (<4%) ризику РСС рекомендація щодо застосування ІКД відповідає класу IIb (можна розглядати, якщо в пацієнта з ГКМП є поширене пізнє накопичення гадолінію [ПНГ] за даними МРТ серця та/або ФВ ЛШ <50%), а рекомендації щодо використання ІКД у пацієнтів із проміжним (від 4 до <6%) і високим (≥6%) ризиком відповідають класам IIb (можна розглядати) і IIa (слід розглядати) відповідно. Рекомендація щодо імплантації ІКД після перенесеної РСС (вторинна профілактика) відповідає класу I (рекомендується) [3]. 2. ДКМП і НДКМП ЛШ. У цих розділах настанови насамперед привертає увагу те значення, яке тепер надають результатам генетичних досліджень та оцінці змін у міокарді за даними МРТ серця в стратифікації ризику РСС. До описаних раніше генних змін, пов’язаних із високим ризиком РСС (FLNC, RBM20, LMNA, PLN), додано гени DSP і TMEM43. Крім того, у настанові описано специфічні ситуації (нез’ясовні непритомності, наявність нестійкої шлуночкової тахікардії [НСШТ], ПНГ), які, за наявності ФВ ЛШ >35%, вимагають вирішення питання про доцільність застосування ІКД. Що стосується НДКМП ЛШ, робоча група припускає, що імплантація ІКД може розглядатися і у випадках із негативним результатом генетичного дослідження за наявності НСШТ, сімейного анамнезу РСС або поширеного ПНГ за даними МРТ серця [3]. 3. АКМП ПШ. При АКМП ПШ, спричиненій шлуночковими аритміями (шлуночкова екстрасистола, НСШТ або шлуночкова тахікардія), антиаритмічними препаратами першого вибору є БАБ (рекомендація класу I). При нездатності БАБ контролювати симптоми, спричинені аритмією, рекомендується розглядати можливість застосування аміодарону (IIa, C) або додавання до БАБ флекаїніду (IIa, C). Для полегшення процесу ухвалення рішень, пов’язаних із первинною профілактикою РСС, рекомендується використовувати стандартні фактори ризику (наявність НСШТ, ФВ ЛШ <45 %, ФВ ПШ <40 %), а також оновлений у 2019 році калькулятор ризику при АКМП ПШ [3]. 4. РКМП. Для випадків РКМП підкреслюється важливість пошуку першопричини з метою проведення етіологічної терапії. Найважливішим методом такого обстеження названо ендоміокардіальну біопсію. Не менш важливо використовувати методи мультимодальної візуалізації, щоб віддиференціювати РКМП від ГКМП і ДКМП із рестриктивною фізіологією. Застосування ІКД рекомендується тільки як вторинна профілактика РСС, аналогічна рекомендація для первинної профілактики відсутня [3]. З повним текстом настанови англійською мовою можна ознайомитися на офіційному сайті European Society of Cardiology за адресою https://www.escardio.org/.
Список літератури знаходиться в редакції
Автор огляду Віктор Мицьо Medicine Review 2024; 3 (76): 30 |
Корисні посилання






|
|
Інформація, розміщена на сайті, призначена тільки для професіоналів охорони здоров'я та не може бути використана як інструкція для самолікування. |
Головна | Про видання | Поточний номер | Архів номерів | Новини | Правова інформація
Medicine Review © 2008—2025. Усі права захищені.
|
мапа сайту корисні посилання |
