Аритмогенная правожелудочковая кардиомиопатия: падчерица в «семействе» кардиомиопатийКардиомиопатии (КМП) – группа различных по этиологии и патогенезу заболеваний сердца, приводящих к необратимым нарушениям структуры и функции миокарда. Патологические изменения миокарда приводят к прогрессирующему ухудшению насосной функции сердца, развитию сердечной недостаточности (СН) и аритмических нарушений, увеличивая риск смерти пациента. Современные представления о КМП имеют не столь уж длинную историю развития и, по сути, только последние годы отличаются активным изучением данной проблемы и во многих случаях серьезным пересмотром этих представлений. Сам термин «кардиомиопатия» появился еще в 1957 г., и его автор, Wallace Brigden, предложил его для обозначения первичных заболеваний миокарда неизвестной этиологии [1]. С тех пор представления о КМП постепенно расширялись, но наиболее значительный прорыв в понимании заболеваний этого рода сделан только в последние 10-20 лет, когда были выделены отдельные формы КМП, получено много важной информации об их этиологии и патогенезе, проведен ряд клинических исследований по изучению преимуществ различных стратегий лечения при КМП. В 1995 г. Всемирной организацией здравоохранения (ВОЗ) в соавторстве с экспертами Международного общества и федерации кардиологов (International Society and Federation of Cardiology, ISFC) была разработана классификация КМП [2], которая впервые отказалась от ранее принятого подхода объединять под термином «кардиомиопатии» все заболевания миокарда неизвестной или неясной этиологии. Ранее эксперты ВОЗ разделяли болезни миокарда на так называемые специфические заболевания и на кардиомиопатии (Report of the WHO/ISFC Task Force, 1980). После уточнения неизвестных ранее причин развития КМП и по мере постепенного стирания границ между «специфическими заболеваниями миокарда» и «кардиомиопатиями» стала очевидной необходимость пересмотра номенклатуры и классификации болезней миокарда. В классификации ВОЗ 1995 г. впервые было определено, что КМП – это любые заболевания миокарда, связанные с нарушением его функции. Под этим термином стали объединять все многообразие первичных поражений миокарда. Если этиология и патогенез КМП известны, то она относится к «специфическим кардиомиопатиям». К последним принадлежат ишемическая, клапанная, гипертензивная, воспалительная, метаболическая КМП, изменения миокарда при системных заболеваниях (системной красной волчанке, ревматоидном артрите, склеродермии, саркоидозе, лейкемии и др.), мышечных дистрофиях, нейромышечных нарушениях, токсические и аллергические поражения миокарда, а также КМП беременных. В зависимости от патологоанатомических и патофизиологических особенностей развития КМП были определены четыре основные формы КМП: дилатационная, рестриктивная, гипертрофическая и аритмогенная правожелудочковая. Если КМП невозможно было отнести ни к одной из них, их относили к немногочисленной подгруппе «неклассифицируемых КМП». В 2006 г. Американская ассоциация сердца (American Heart Association, AHA) опубликовала научное соглашение с новой версией классификации КМП [3]. Эта классификация более подробная и, как считают многие специалисты, более корректная, хотя и более сложная. Она основывается на результатах самых современных экспериментальных и клинических исследований, в том числе молекулярно-генетических, и отражает тот факт, что изучение КМП на серьезной научной основе в последние годы только началось. Вкратце следует сказать, что эксперты AHA выделили две большие группы КМП – первичные (наследственные, приобретенные и смешанного происхождения) и вторичные. В этих двух классификациях особое внимание обращает на себя аритмогенная правожелудочковая КМП (АП-КМП). Она является одной из четырех основных форм КМП (по классификации ВОЗ/ISFC, 1995), но, хотя дилатационная, рестриктивная и гипертрофическая КМП достаточно хорошо известны практическим врачам, об АП-КМП вспоминают редко, а имеющиеся о ней знания, как правило, скудны. В частности, в Украине это заболевание практически не диагностируется. Наш обзор посвящен современным представлениям об этом варианте КМП. Определение АП-КМП характеризуется прогрессирующим замещением миокарда правого желудочка (ПЖ) жировой и фиброзной тканью, с частичным вовлечением левого желудочка (ЛЖ) и межжелудочковой перегородки. Это особое заболевание, которое, скорее всего, имеет генетическую природу; хотя, возможно, АП-КМП не является однородной патологией и объединяет некоторую совокупность схожих заболеваний. АП-КМП выделили как самостоятельную болезнь совсем недавно, около 20 лет назад. В классификации AHA (2006) АП-КМП относится к первичным КМП, имеющим наследственное происхождение. Следует обратить особое внимание на то, что в отечественной литературе встречаются разночтения термина «аритмогенная кардиомиопатия», и ряд авторов под АП-КМП понимает вторичные изменения миокарда с развитием хронической сердечной недостаточности (СН), возникшие в результате длительно существующей аритмии (как правило, вследствие фибрилляции предсердий). Это неправильное использование данного термина, и мы рекомендуем практическим врачам ориентироваться прежде всего на терминологию ВОЗ/ISFC и AHA. Эксперты ВОЗ и AHA используют термин «аритмогенная правожелудочковая кардиомиопатия» по отношению к определенному и достаточно подробно изученному заболеванию, при котором изменения миокарда первичны (и, по всей видимости, генетически обусловлены), а аритмические нарушения развиваются на их основе, то есть вторично (и чаще всего это желудочковые нарушения ритма, а не предсердные). Именно это заболевание подразумевается в классификациях ВОЗ/ISFC 1995 г. [2] и AHA 2006 г. [3]. Нарушения сердечного ритма, особенно тахисистолическая форма мерцательной аритмии действительно может значительно усугублять дисфункцию желудочков и даже самостоятельно вызывать ее без выраженного предшествовавшего органического поражения миокарда, однако такой вариант сочетания аритмии и СН не следует называть аритмогенной кардиомиопатией. Эпидемиология АП-КМП считается относительно редким заболеванием, тем не менее эта патология заслуживает особого внимания, поскольку она является одной из ведущих причин развития внезапной сердечной смерти среди молодых людей (особенно среди спортсменов). Распространенность АП-КМП в общей популяции оценивается по разным данным от 1:3000 до 1:10 000 (в среднем приблизительно 1:5000, согласно научному соглашению AHA, 2006 [3]). Некоторые авторы докладывают о более высокой встречаемости АП-КМП в отдельных регионах (1:2000-1:2500) – например, в Италии («венецианская кардиомиопатия»), Греции («болезнь Наксоса») [6, 9, 11, 12, 15]. Большинство больных – мужского пола (1:3-1:4). Многие авторы считают, что истинная распространенность этого заболевания может быть гораздо выше, но значительная часть случаев не распознается ни клинически, ни патологоанатомически и остается недиагностированной. В ряде стран мира (в том числе и в Украине) диагностика этой КМП вообще практически не проводится, но это не обозначает, что таких больных нет или что их количество в данном регионе ничтожно мало. Актуальность АП-КМП достаточно высока, учитывая то, что она в настоящее время признана одной из известных причин внезапной сердечной смерти у молодых людей. Летальность при этой патологии составляет 2-4% в год [16]. Существуют основания полагать, что, по крайней мере, в Италии АП-КМП является одной из основных причин внезапной сердечной смерти у спортсменов (G. Thiene et al., 1988; B.J. Maron, 1988). На рисунке 1 показано распределение основных причин внезапной сердечной смерти у молодых людей в Северной Италии [9]. По данным G. Thiene et al. (1988), АП-КМП находится на втором месте, обусловливая 13% всех случаев внезапной сердечной смерти.
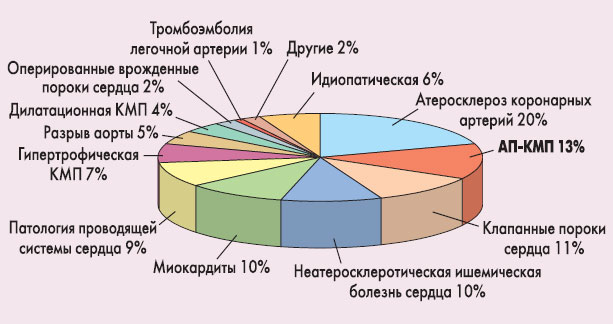 Рисунок 1. Основные причины внезапной сердечной смерти у молодых людей в Северной Италии (по данным G. Thiene et al., 1988)
Однако справедливости ради следует отметить, что в большинстве других исследований, посвященных изучению вероятных причин внезапной сердечной смерти у молодых лиц, АП-КМП в этом отношении уступала первенство многим другим причинам, таким как врожденные аномалии коронарных артерий, гипертрофическая КМП, синдром Марфана, пролапс митрального клапана, стеноз клапана аорты и др. Этиология Недостаточно четкое представление о природе АП-КМП связано с тем, что, хотя заболевание имеет явно семейный характер, оно проявляется по наследственной линии не всегда. Кроме того, эта форма КМП часто (до 75% случаев) ассоциирована с миокардитом (обусловленным энтеровирусом, аденовирусом или др.), что долго заставляло ученых думать о возможном приобретенном происхождении патологии. В настоящее время принято считать, что для АП-КМП характерен аутосомно-доминантный тип наследования с неполной пенетрантностью гена. Это обозначает, что заболевание развивается только у части лиц, в генотипе которых присутствует аномальный ген; у остальных носителей доминантного гена наследственное предрасположение к болезни остается нереализованным. Однако описан также, по крайней мере, один вариант АП-КМП, который наследуется по аутосомно-рецессивному типу («болезнь Наксоса»). Изучение генетической природы АП-КМП остается одним из наиболее активных направлений в молекулярно-генетической кардиологии. Тесная ассоциация с миокардитом продолжает исследоваться, но большинство авторов предполагают, что миокардит появляется вторично, в связи с перерождением миокарда и изменением его восприимчивости к инфекциям и реактивности ткани. Патогенез и клинические проявления При АП-КМП миокард ПЖ начинает прогрессивно терять миоциты, которые замещаются жировой или фиброзно-жировой тканью. На ранних стадиях такого перерождения миокарда стенки ПЖ утолщаются, но в дальнейшем напротив – истончаются, в них появляются небольшие аневризмы. Сначала эти изменения имеют региональный, ограниченный характер, затем постепенно распространяются на весь ПЖ, в большинстве случаев захватывая частично ЛЖ (по данным D. Corrado et al., 1997, более чем у половины больных, хотя обычно поражение ЛЖ ограничивается небольшим субэпикардиальным участком в заднебоковой стенке желудочка и только в редких случаях имеет выраженный характер, приводя к аневризмам стенки ЛЖ и его дилатации) и межжелудочковую перегородку (это бывает редко и в незначительной степени, потому что перерождение начинается от эпикарда, а межжелудочковая перегородка не имеет эпикарда). Некоторое время заболевание протекает бессимптомно. Но поскольку очаги фиброзно-жирового перерождения не проводят электрические импульсы, по мере прогрессирования этого перерождения и увеличения таких очагов электрическая активность сердца становится все более беспорядочной, в связи с чем развиваются нарушения ритма сердца и его сократимости. Ухудшение насосной функции ПЖ ведет к расширению камер правого сердца, систолической дисфункции, СН. Первые клинические проявления обычно возникают у молодых людей (до 40 лет). Как правило, в клинике АП-КМП проявляется в первую очередь желудочковыми аритмиями (часто мономорфной желудочковой тахикардией, у многих больных также встречаются желудочковая экстрасистолия, эпизоды фибрилляции желудочков, реже мерцание или трепетание предсердий), в связи с чем патология и получила такое название. В типичных случаях пациент жалуется на приступы сердцебиения, тахикардии, частые головокружения и обмороки. Однако аритмические нарушения не являются единственным или специфическим симптомокомплексом при АП-КМП. Как и при любой другой КМП, у этих пациентов имеются и многие другие проблемы, прежде всего те, которые связаны с СН (изолированной правожелудочковой или бивентрикулярной). В результате клиническая картина при АП-КМП может быть достаточно разнообразной. В ряде случаев первым клинически значимым проявлением АП-КМП выступает внезапная сердечная смерть (вследствие фибрилляции желудочков) – как правило, во время физических нагрузок, при занятиях спортом. Особенности ведения пациента с АП-КМП
Диагностика Диагностика этого заболевания может быть достаточно сложной задачей, учитывая его невысокую распространенность и отсутствие специфических признаков. Обычно заподозрить АП-КМП можно только оценив всю совокупность анамнестических, клинических, электрокардиографических, эхокардиографических, рентгенографических и других визуализирующих методов исследования и исключив более вероятные формы патологии миокарда (миокардит, воспалительную КМП, дилатационную КМП и др.). Очень характерными для АП-КМП являются аритмии, синкопе, эпизоды внезапной остановки сердца в анамнезе. При неинвазивных визуализирующих методах обследования на вероятность наличия у пациента АП-КМП может указывать расширение камер правого сердца и/или аномальные движения стенки ПЖ, нарушение сократимости правого желудочка (асинергия, гипокинезия), аневризмы ПЖ. По данным магнитно-резонансной томографии можно обнаружить участки замещения миокарда жировой тканью, истончение стенок ПЖ, аневризмы; последние научные данные свидетельствуют также о перспективности усиления магнитно-резонансного сигнала с помощью контрастирования гадолинием. Нарушения сократимости ПЖ, его дилатация и аневризмы визуализируются также с помощью рентгенконтрастной вентрикулографии. В последние годы изучаются возможности нового инвазивного метода диагностики – трехмерного электроанатомического картирования, который позволяет отличить фиброзно-жировое перерождение от воспалительных изменений, что очень важно, так как АП-КМП часто сложно дифференцировать от воспалительной КМП. В свете дифференциальной диагностики АП-КМП следует подчеркнуть, что для этой патологии весьма характерна очаговость поражения стенки ПЖ. Обычно только на поздних стадиях очаги сливаются настолько, что поражение ПЖ приобретает характер диффузного. Это в наибольшей степени отличает АП-КМП от правожелудочковой дилатационной КМП и миокардита, при которых гипокинезия ПЖ тотальная. Обнаружение отдельных участков гипокинезии, утончения стенки ПЖ и особенно – аневризм ПЖ (прежде всего у молодого пациента, особенно если у него есть обмороки, приступы сердцебиения, аритмические нарушения в анамнезе) должно насторожить врача в отношении возможной АП-КМП. Точный диагноз подтверждается с помощью эндомиокардиальной биопсии свободной стенки ПЖ. Гистологическое исследование выявляет фиброзно-жировую инфильтрацию миокарда ПЖ, атрофию мышечной ткани, иногда видны разрушающиеся кардиомиоциты в окружении воспалительных инфильтратов. Однако главная проблема при этом состоит в том, что поражение миокарда при АП-КМП очаговое, и забор материала может быть произведен из интактных участков. Кроме того, поскольку фиброзно-жировое перерождение при АП-КМП распространяется по направлению от эпикарда к эндокарду, эндомиокардиальная биопсия может не захватить гистологически измененную ткань даже в области очага перерождения (если оно еще не достигло эндокарда). В 1994 г. эксперты Европейского общества кардиологов (European Society of Cardiology, ESC) и ISFC предложили следующие критерии для диагностики АП-КМП [4]. Большие диагностические критерии:
Малые диагностические критерии:
Для диагностирования АП-КМП достаточно сочетания двух больших критериев, либо одного большого и двух малых, либо четырех малых критериев. M.S. Hamid et al. в 2002 г. также выдвинули предложение о том, чтобы большинство из малых критериев по отдельности также рассматривались как основание для диагностики АП-КМП у пациентов, являющихся ближайшими родственниками лиц, у которых АП-КМП диагностирована ранее [5]. Это помогает обнаружить патологию на ранних стадиях или выявить лиц с неполной фенотипической экспрессией гена. Диагностика может быть весьма затруднена, если АП-КМП не обусловливает аритмии и/или ассоциирована с миокардитом, а также при достаточно диффузном поражении, при котором обычно ошибочно подозревается дилатационная КМП. Следует помнить и о таких вариантах течения этой патологии.
Прогноз Молодой возраст пациента, случаи внезапной сердечной смерти в анамнезе, выраженная и плохо переносимая желудочковая тахикардия (особенно полиморфная), частые эпизоды синкопе, тяжелая дисфункция ПЖ, СН (особенно с вовлечением систолической функции ЛЖ), наличие в семье родственников, умерших в раннем возрасте предположительно от внезапной сердечной смерти, – все эти факторы являются предикторами неблагоприятного прогноза при АП-КМП. Возможности стратификации риска при этой патологии продолжают изучаться.
Лечение К сожалению, в настоящее время не найдены способы замедления или остановки прогрессирования перерождения миокарда при АП-КМП. Ведение пациента с этим заболеванием должно подразумевать рекомендации по модификации образа жизни (помимо стандартных кардиопротективных мероприятий, больному следует избегать чрезмерных физических нагрузок, даже при асимптомной АП-КМП), лечение аритмических нарушений и СН, профилактику внезапной сердечной смерти. Снижение риска внезапной сердечной смерти является одной из основных задач лечения АП-КМП. Своевременное назначение адекватной медикаментозной терапии (β-блокаторами, противоаритмическими средствами), проведение абляции атриовентрикулярного узла, имплантация кардиовертера-дефибриллятора (ИКД) позволяют существенно снизить риск этого осложнения, часто фатального. Одним из наиболее показанных лечебных подходов при АП-КМП является ИКД. Кардиовертер-дефибриллятор эффективно предупреждает развитие внезапной сердечной смерти у данной категории больных, а также уменьшает прогрессирование сократительной дисфункции миокарда и снижает риск развития СН. Исследования D. Corrado et al. (2003), T. Wichter et al. (2004), A. Roguin et al. (2004) убедительно продемонстрировали, что ИКД улучшает долгосрочный прогноз у пациентов высокого риска с АП-КМП. Наилучшими кандидатами для ИКД являются пациенты высокого риска – с эпизодами остановки сердца в анамнезе, с гемодинамически значимой желудочковой тахикардией, с вовлечением в патологический процесс ЛЖ, с частыми необъяснимыми синкопе. У этих больных ИКД в течение 36 месяцев обеспечивает снижение летальности на 24-35% (D. Corrado et al., 2003; T. Wichter et al., 2004; A. Roguin et al., 2004). У пациентов с хорошо переносимыми и нежизнеугрожающими нарушениями сердечного ритма, не влияющими на гемодинамику, долгосрочный прогноз гораздо лучше, поэтому у них в качестве терапии первой линии более рационально использование антиаритмических препаратов и β-блокаторов. Современная доказательная база позволяет утверждать, что наиболее эффективными противоаритмическими препаратами у данной категории больных являются соталол и амиодарон, которые применяются в качестве монотерапии или в комбинации с β-блокаторами. Соталол продемонстрировал самую высокую эффективность по сравнению с другими препаратами и потому считается препаратом выбора (T. Wichter et al., 1992); амиодарон показан в случае непереносимости соталола или отсутствия ответа на него. Однако способность такого лечения снижать риск внезапной сердечной смерти остается недоказанной. Часто практикуется также абляция атриовентрикулярного узла, но следует отметить, что после этого вмешательства в большей части случаев (до 85% по некоторым данным) желудочковая тахикардия со временем рецидивирует, что связано с появлением новых аритмогенных зон в результате прогрессирования фиброзно-жирового перерождения миокарда (D. Dalal et al., 2007). Через 3 года после абляции доля пациентов, у которых не было рецидива аритмии, составляет не более 40%. Поэтому абляция обычно приберегается как метод лечения второй линии и используется при рефрактерности аритмии к медикаментозной терапии, при частом рецидивировании желудочковой тахикардии после ИКД. Вместе с тем долгосрочная выживаемость больных после абляции улучшается (T. Wichter et al., 2005). В случае развившейся СН необходимы стандартные лечебные мероприятия – применение диуретиков, ингибиторов АПФ, дигоксина, антикоагулянтов. Острая СН, возникшая на фоне приступа тяжелой аритмии, требует госпитализации в стационар, введения инотропных средств и других подходов по стабилизации гемодинамики пациента. В этом отношении для лечения осложнений, развившихся на фоне АП-КМП, наиболее интересны препараты, которые, помимо основного действия, также будут бороться с нарушениями сердечного ритма или, по крайней мере, не имеют выраженного проаритмогенного эффекта, свойственного многим препаратам кардиологической группы. Так, хотелось бы отметить такой инотропный препарат, как левосимендан. Он повышает чувствительность сократительных белков миокарда к ионам кальция, что не только обусловливает увеличение силы сердечных сокращений, но и улучшает внутрисердечную проводимость. Препарат пока не изучался целенаправленно у больных с АП-КМП, однако продемонстрировал положительные результаты при лечения декомпенсированной СН, развившейся на фоне других КМП – дилатационной, ишемической, КМП беременных. Например, в двойном-слепом плацебо-контролированном исследовании LIDO (2002) на фоне использования левосимендана у больных с СН было гораздо меньше случаев нарушений частоты и ритма сердечных сокращений (мерцательной аритмии, экстрасистолий, фибрилляции желудочков, брадикардии), чем при применении добутамина (3,9 vs 13%, р=0,023). Это, наряду с улучшением гемодинамики, отразилось на различиях в уровне смертности на фоне приема левосимендана и добутамина: 8 vs 17% соответственно (р=0,049) в течение 31 суток (относительное снижение риска смерти на 57%) и 26 vs 38% соответственно (р=0,029) в течение 180 суток (относительное снижение риска смерти на 43%). Важно отметить также, что гемодинамическая эффективность левосимендана не ослабляется на фоне применения β-блокаторов (в отличие от добутамина), что может быть принципиально важным, учитывая, что β-блокаторы являются одной из базисных групп препаратов, показанных при АП-КМП. Более традиционные инотропы, широко использующиеся в клинической практике, имеют проаритмогенный эффект и потому могут оказаться не самым удачным выбором для лечения СН, основой для которой стала АП-КМП. Тяжелые случаи АП-КМП с некурабельной желудочковой аритмией и тяжелой СН делают пациента кандидатом для трансплантации сердца [14]. Лечение целесообразно только в случае клинически явной АП-КМП; у асимптомных больных и у носителей гена без заболевания нет необходимости в таких мероприятиях – во всяком случае, эффективность профилактики внезапной сердечной смерти с помощью приема β-блокаторов и других методов лечения у данной категории пациентов должна быть изучена в соответствующих клинических исследованиях. Асимптомные больные должны регулярно обследоваться у кардиолога, и начинать получать лечение при появлении первых симптомов (аритмии, СН и др.). Однако некоторые авторы рекомендуют назначать таким пациентам β-блокаторы [9]. Заключение Большинство исследований, касающихся АП-КМП, – небольшие, нерандомизированные, в ряде случаев имеющаяся информация об этой патологии почерпнута из отдельных клинических случаев. Генетические изыскания в этой области ведутся очень активно, однако пока они не проясняют всех противоречий, связанных с этим заболеванием. Множество пробелов в наших знаниях об АП-КМП объясняются и относительной редкостью встречаемости заболевания, и сложностью его диагностики, особенно на доклиническом (асимптомном) этапе, и неоднозначным этиопатогенезом этой патологии, и тем, что во многих случаях, вероятно, АП-КМП не распознается даже посмертно. Ученые предполагают, что истинная распространенность АП-КМП может быть гораздо выше, чем принято считать в настоящее время, особенно учитывая то, что первым проявлением этого заболевания зачастую бывает внезапная сердечная смерть, которую часто расценивают как идиопатическую, с неизвестной этиологией. В какой-то доле случаев причиной внезапной сердечной смерти у молодого человека может выступать именно недиагностированная АП-КМП. Это заболевание известно не так давно, и к настоящему времени ученые уже достаточно серьезно продвинулись в его изучении, однако впереди еще множество нерешенных вопросов. Для получения ответов на эти вопросы несколько лет назад стартовали две крупные исследовательские программы по изучению АП-КМП – в США (F. Marcus et al., 2003) и в Европе (C. Basso et al., 2004); обсуждается необходимость создания единого международного регистра этого заболевания. Объединение данных из многих стран мира позволит существенно продвинуться в понимании этой сложной патологии, поэтому хотелось бы обратить внимание и украинских врачей на эту малоизученную, но незаслуженно отодвинутую в сторону проблему. Литература: 1. Brigden W. Uncommon myocardial disease. The non-coronary cardiomyopathies. Lancet 1957; 273 (7007): 1179-84. 2. Richardson P., McKenna W., Bristow M. et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996; 93 (5): 841-2. 3. Maron B.J., Towbin J.A., Thiene G. et al.; American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113 (14): 1807-16. 4. McKenna W.J., Thiene G., Nava A. et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J 1994; 71 (3): 215-8. 5. Hamid M.S., Norman M., Quraishi A. et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol 2002; 40: 1445-50. 6. Corrado D., Basso C., Thiene G. Arrhythmogenic right ventricular cardiomyopathy: an update. Heart 2009; 95: 766-773. 7. Marcus F., Towbin J.A. The mystery of arrhythmogenic right ventricular dysplasia/cardiomyopathy: from observation to mechanistic explanation. Circulation 2006; 114 (17): 1794-5. 8. Pike R. Arrhythmogenic right ventricular cardiomyopathy. Can J Cardiovasc Nurs 2009; 19 (2): 5-9. 9. Thiene G., Corrado D., Basso C. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Orphanet J Rare Dis 2007; 2: 45. 10. Wichter T., Paul T.M., Eckardt L. et al. Arrhythmogenic right ventricular cardiomyopathy. Antiarrhythmic drugs, catheter ablation, or ICD? Herz 2005; 30 (2): 91-101. 11. Corrado D., Buja G., Basso C., Thiene G. Clinical diagnosis and management strategies in arrhythmogenic right ventricular cardiomyopathy. J Electrocardiol 2000; 33 Suppl: 49-55. 12. Corrado D., Basso C., Nava A., Thiene G. Arrhythmogenic right ventricular cardiomyopathy: current diagnostic and management strategies. Cardiol Rev 2001; 9 (5): 259-65. 13. Frances R.J. Arrhythmogenic right ventricular dysplasia/ cardiomyopathy. A review and update. Int J Cardiol 2006; 110 (3): 279-87. 14. Fiorelli A.I., Coelho G.H., Oliveira J.L. Jr. et al. Heart transplantation in arrhythmogenic right ventricular dysplasia: case reports. Transplant Proc 2009; 41 (3): 962-4. 15. Basso C., Corrado D., Marcus F.I. et al. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009; 373 (9671): 1289300. 16. Herren T., Gerber P.A., Duru F. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: a not so rare «disease of the desmosome» with multiple clinical presentations. Clin Res Cardiol 2009; 98 (3): 141-58. 17. Anderson E.L. Arrhythmogenic right ventricular dysplasia. Am Fam Physician 2006; 73 (8): 1391-8. 18. Sen-Chowdhry S., Lowe M.D., Sporton S.C., McKenna W.J. Arrhythmogenic right ventricular cardiomyopathy: clinical presentation, diagnosis, and management. Am J Med 2004; 117 (9): 685-95. 19. Kies P., Bootsma M., Bax J. et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: screening, diagnosis, and treatment. Heart Rhythm 2006; 3 (2): 225-34. 20. El Demellawy D., Nasr A., Alowami S. An updated review on the clinicopathologic aspects of arrhythmogenic right ventricular cardiomyopathy. Am J Forensic Med Pathol 2009; 30 (1): 78-83. Автор обзора Анна Карташева Medicine Review 2009; 3 (08): 46-51 |
Корисні посилання






|
|
Інформація, розміщена на сайті, призначена тільки для професіоналів охорони здоров'я та не може бути використана як інструкція для самолікування. |
Головна | Про видання | Поточний номер | Архів номерів | Новини | Правова інформація
Medicine Review © 2008—2026. Усі права захищені.
|
мапа сайту корисні посилання |
